
Прогрессирующая оссифицирующая фибродисплазия
Прогрессирующая оссифицирующая фибродисплазия – тяжелое генетическое заболевание, характеризующееся нарушением обмена в тканях мышц, сухожилий и связок, что приводит к образованию в них кальцинатов. Симптомами этого состояния являются уплотнения в мягких тканях шеи, спины и конечностей, мышечная гипотония, в дальнейшем – выраженные деформации позвоночника, скованность движений вплоть до инвалидизации больных. Диагностика прогрессирующей оссифицирующей фибродисплазии производится на основании данных настоящего статуса пациента, рентгенологических и молекулярно-генетических исследований. Специфического лечения патологии на сегодняшний день не существует, но имеются обнадеживающие результаты опытов на клеточных культурах больных данным заболеванием.
Общие сведения
Прогрессирующая оссифицирующая фибродисплазия (ФОП, параоссальная гетеротопическая оссификация) – редкое генетическое заболевание, при котором возникают процессы патологического окостенения сухожилий, фасций, межмышечных перегородок и других структур, состоящих из волокнистой соединительной ткани. Впервые это состояние было упомянуто французским врачом Гаем Петимом еще в 1692 году, однако его наиболее полное описание и изучение произвел в 70-х годах XX века американский врач-генетик Виктор Маккьюзик, который и дал название этому заболеванию. Прогрессирующая оссифицирующая фибродисплазия чаще всего является результатом спонтанных герминативных мутаций, однако описаны и семейные случаи этой патологии, при этом она характеризуется аутосомно-доминантным механизмом передачи со 100% пенетрантностью. Встречаемость оценивается примерно в 1:1 800 000-2 000 000, заболевание с равной долей вероятности поражает как мальчиков, так и девочек. Характерной особенностью прогрессирующей оссифицирующей фибродисплазии является тот факт, что любые попытки оперативного лечения этого состояния (устранение очагов оссификации или облегчение движений в суставах) приводят к взрывному росту новых гетеротопических участков костной ткани.
Причины прогрессирующей оссифицирующей фибродисплазии
Этиология прогрессирующей оссифицирующей фибродисплазии заключается в нарушении регуляции процессов формирования костной ткани. В эмбриональном периоде за развитие костей отвечает ряд рецепторов, работа которых приводит к образованию твердой соединительной ткани в соответствующих областях тела. При развитии этого заболевания возникает мутация гена ACVR1, расположенного на 2 хромосоме. Продуктом экспрессии данного гена является специфический белок – активин-рецептор 1 типа. Он относится к более обширной группе BMP-рецепторов, которые обладают способностью связывать вещества-факторы роста костной ткани и передавать соответствующие сигналы внутрь клеток. Чаще всего причиной прогрессирующей оссифицирующей фибродисплазии являются миссенс-мутации (например, Arg206His) в 6 экзоне гена ACVR1.
В результате этого генетического нарушения происходит образование дефектной формы активина-рецептора 1 типа, который начинает образовываться не только в эмбриональном периоде и в остеобластах у взрослого человека, но и в клетках волокнистой соединительной ткани. Дальнейший патогенез прогрессирующей оссифицирующей фибродисплазии изучен недостаточно по причине редкости этого состояния. Однако большинство исследователей склоняются к мнению, что дефектный рецептор становится способным реагировать не только на соединения из группы BMP (bone morphogenetic protein – белки костного морфогенеза), но и на другие вещества – в частности, факторы воспаления. Доказательством этой теории служит тот факт, что очаги гетеротопической оссификации чаще всего возникают на месте повреждения мягких тканей – ушибов, порезов, укусов насекомых. Кроме того, при гистологическом изучении патологических уплотнений в них всегда наблюдается умеренная лимфоцитарная инфильтрация, что указывает на воспалительный или иммунный характер процесса.
Мутации гена ACVR1 приводят не только к аномальному костному морфогенезу у детей и взрослых, они также нередко обуславливают ряд врожденных пороков развития опорно-двигательного аппарата. Наиболее распространенными проявлениями прогрессирующей оссифицирующей фибродисплазии такого типа являются микродактилии, варусное искривление большого пальца стопы (клинодактилия). Из других, более редких костных аномалий, отмечают сращение тел шейных позвонков, дисплазии метафизов, сращение реберно-позвоночных суставов. По причине сочетания дисплазии костной ткани и слабости мышечного корсета спины у больных часто развиваются тяжелые искривления позвоночника, что приводит к вторичным неврологическим нарушениям.
Симптомы прогрессирующей оссифицирующей фибродисплазии
При рождении прогрессирующая оссифицирующая фибродисплазия может проявлять себя лишь нарушениями формирования костей пальцев рук и ног, никаких других изменений при этом не обнаруживается. Начало заболевания внезапное, может произойти без всяких видимых причин в возрасте от нескольких месяцев до 3-5 лет. Обычно первыми симптомами данной патологии становятся уплотнения в мышечных тканях шеи, спины и рук, размеры которых могут составлять от 1 до 10 сантиметров. При пальпации очаги бывают болезненными, кожа над ними либо не изменена, либо (в редких случаях) гиперемирована. При прогрессирующей оссифицирующей фибродисплазии такие уплотнения могут появляться на месте ушибов, порезов и других травм мягких тканей.
Походка больных прогрессирующей оссифицирующей фибродисплазией, как правило, характеризуется скованностью, малыми шагами. Лицо амимичное на фоне напряженных мышц шеи и спины. По мере нарастания количества и размеров уплотнений объем движений в суставах постепенно уменьшается, приводя на терминальных этапах заболевания практически к полному обездвиживанию. Нередко отмечаются боли неврологического характера, обусловленные сдавлением корешков спинномозговых нервов, также могут поступать жалобы на нарушение кожной чувствительности. Все эти проявления прогрессирующей оссифицирующей фибродисплазии постепенно нарастают и становятся все более выраженными.
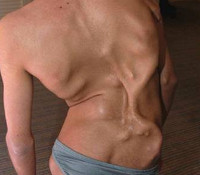
Описаны случаи, когда очаги на первоначальном этапе развития самопроизвольно рассасываются (в том числе и под воздействием некоторых лекарственных средств), однако в последующем уплотнения неизменно возникают снова. В течение 2-3 недель происходит оссификация, после чего их самопроизвольное или терапевтическое разрешение становится невозможным. У взрослых больных прогрессирующей оссифицирующей фибродисплазией выявляются десятки таких очагов, которые могут сливаться между собой, образуя картину «второго скелета». Это обстоятельство наряду с многочисленными деформациями костей приводит к глубокой инвалидизации больных и нередко провоцирует развитие многочисленных нарушений со стороны внутренних органов с последующим летальным исходом.
Диагностика и лечение прогрессирующей оссифицирующей фибродисплазии
Диагностика прогрессирующей оссифицирующей фибродисплазии осуществляется на основании данных осмотра пациента, рентгенологических и генетических исследований. Как правило, результаты осмотра больных зависят от возраста и выраженности заболевания. У детей до 10-12 лет основными симптомами являются очаги уплотнения мягких тканей на голове, шее, иногда на спине. У пациентов старшего возраста очаги оссификации могут располагаться на любом участке тела, нередко отмечается резкое ограничение подвижности суставов, выраженные искривления позвоночника. Уплотнения мягких тканей приводят к появлению характерных бугров на коже и зачастую сильно уродуют больного прогрессирующей оссифицирующей фибродисплазией. Иногда в качестве дополнительного метода диагностики прибегают к биопсии и последующему гистологическому исследованию очагов уплотнения, однако многие специалисты выступают против этой техники, поскольку такая инвазивная процедура способна спровоцировать развитие нового очага оссификации.
Рентгенологическое исследование на ранних этапах заболевания может обнаруживать врожденные пороки развития опорно-двигательного аппарата – клинодактилию больших пальцев ног, дисплазию метафизов, укорочение длинных трубчатых костей конечностей. По мере развития гетеротопической оссификации в сухожилиях, фасциях, межмышечных соединительнотканных перегородках выявляются сначала одиночные, а затем и множественные тени, имеющие костную плотность. В далеко зашедших случаях прогрессирующей оссифицирующей фибродисплазии тени сливаются между собой и нередко затрудняют визуализацию внутренних органов и других глубоко расположенных структур. При помощи методов современной генетики можно диагностировать это состояние путем поиска мутаций в гене ACVR1.
Специфического лечения прогрессирующей оссифицирующей фибродисплазии не существует, а паллиативная терапия резко ограничена в своих возможностях по причине детского возраста большинства пациентов и специфической реакции организма. Многочисленные попытки устранить очаги гетеротопической оссификации хирургическим путем заканчивались неудачно – после операции из-за повреждения тканей рост уплотнений резко усиливался. На ранних этапах заболевания немного затормозить развитие патологических очагов можно при помощи высоких доз кортикостероидных препаратов и других средств, угнетающих воспалительные процессы (НПВС, ингибиторы лейкотриенов, блокаторы мастоцитов). В последние годы получены первые лабораторные данные по поводу специфического лечения прогрессирующей оссифицирующей фибродисплазии – при помощи особой РНК удалось ингибировать экспрессию дефектного гена ACVR1, не затрагивая его здоровую гомологичную копию в стволовых клетках больных. Возможно, в дальнейшем это позволит с успехом лечить пациентов, страдающих данным заболеванием, или, как минимум, значительно повысить их качество жизни.
Прогноз и профилактика прогрессирующей оссифицирующей фибродисплазии
Прогноз прогрессирующей оссифицирующей фибродисплазии крайне неблагоприятный – проявления заболевания неуклонно нарастают, эктопические очаги оссификации становятся крупнее и сливаются между собой, деформации позвоночника практически лишают больного подвижности. Как правило, летальный исход наступает в 25-40 лет от нарушений дыхания, пневмонии, других расстройств со стороны внутренних органов. Описаны случаи развития очагов оссификации в жевательной мускулатуре, что приводило к невозможности открыть рот и требовало срочных мер по организации питания больного. Поддерживающее лечение при прогрессирующей оссифицирующей фибродисплазии теоретически может замедлить развитие патологии, но на практике его эффективность очень сильно отличается у разных больных. Каких-либо методов профилактики этого редкого генетического заболевания на сегодняшний день не разработано.
Прогрессирующая оссифицирующая фибродисплазия (ФОП)
Фибродисплазия относится к редчайшему заболеванию, которое возникает в результате мутации определенного гена. На сегодняшний день в мире зарегистрировано 600 человек, страдающих от него. Патология характеризуется окостенением мышц, связок, сухожилий. Сначала поражается позвоночник, далее страдают другие кости организма.
Характеристика заболевания
Оссифицирующая фибродисплазия (ФОП) по МКБ 10 имеет номер М61.1, возникает из-за генетических мутаций, в результате видоизменения гена ACVR1. Известны случаи, когда поражаются несколько членов семьи, которые находятся в кровном родстве, братья и сестры. Встречается передача заболевания от одного поколения к другому, от отцов к детям.
Данная патология носит название болезни «второго скелета». Прогрессирующую фибродисплазию провоцируют воспалительные процессы мышц, сухожилий, связок, мягких тканей, по истечении времени приводя к окостенению.

Не следует путать данное заболевание с фиброзной дисплазией, при котором происходит замещение здоровой ткани кости на включение костной трабекулы. Фиброзная дисплазия относится к разряду опухолевых состояний, но не является раковой опухолью. Часто происходит перерождение в доброкачественное новообразование, онкология встречается редко.
У пациентов любой ушиб, небольшая ранка не вылечивается, на месте повреждения происходит образование костной ткани.
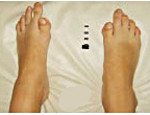
У новорожденного почти полностью определяется наличие заболевания, от которого одинаково страдают и девочки, и мальчики. Оно обнаруживается загнутым внутрь большим пальцем ноги и отсутствием сустава. Патология проявляется в первые 10 лет жизни больного, характеризуется внезапностью. Нижние конечности страдают в последнюю очередь.
Насколько быстро будет развиваться болезнь установить невозможно. Так как это зависит от количества полученных травм и перенесенных оперативных вмешательств.
Бывают случаи, когда врачи путают оссифицирующую фибродисплазию с онкологией, которая приводит к формированию костных образований. После их удаления происходит резкое усугубление ситуации и ускоренное развитие патологии.
Как развивается
Оссифицирующая фибродисплазия развивается по стадиям:
- На первой стадии образуется небольшой воспалительный очаг;
- На второй стадии патология начинает прогрессировать, воспаление вовлекается в ткани, расположенные около воспаленного места;
- На третьей стадии образуются бессосудистые уплотнения;
- Далее формируется хрящеобразование;
- Завершающей стадией является окостенение.
Причины
Оссифицирующая фибродисплазия начинает свое формирование еще в период внутриутробного развития, в результате изменения гена, расположенного во второй хромосоме. Из-за чего происходит мутация формы белка, отвечающего за развитие костной ткани.
Гетеротопические фибродисплазийные очаги обычно формируются в местах повреждения мягкой ткани, ожогов, укусов насекомых, порезов, ушибов. В данной области обнаруживается много лейкоцитов, являющихся фактором иммунного, воспалительного процесса.
Имеются пациенты, которые являются носителями гена, и при определенных обстоятельствах заболевание начинает прогрессировать. На скорость его проникновения оказывает воздействие активность гена. К провоцирующим факторам относятся:
- Хирургические вмешательства;
- Вакцинирование;
- Травмирование кожного покрова;
- Вирусная инфекция

Симптомы
Оссифицирующая фибродисплазия редко начинает развиваться сразу же после рождения ребенка. Врачи обращают внимание на нарушение сформированности пальчиков на ногах и руках. Никаких других внешних проявлений не наблюдается.
Оссификация начинается внезапно, порой без наличия объективной причины. Возраст ребенка может быть от 2-3 месяцев до нескольких лет. Чаще прогрессирование происходит в первые пять лет жизни. Симптомами заболевания считаются:
- Ранняя симптоматика проявляется формированием небольшого уплотнения, размером до 3 мм;
- При прогрессировании уплотнение увеличивается до 10 см;
- При пальпации наблюдается болезненность, иногда покраснение;
- Походка у пациента приобретает неуверенность, скованность;
- На лице отсутствуют какие-либо эмоции;
- Происходит уменьшение объема движений, что имеет связь с появлением новых уплотнений, приводя к полной обездвиженности;
- При прощупывании появляются боли в ногах;
- Нарушается чувствительность;
- Происходят сбои функционирования внутренних органов.
Наиболее очевидной клинической картиной заболевания считают окостенение мягкой ткани. Но возможно проявление следующих дефектов, говорящих о наличии оссифицирующей фибродисплазии:
- Короткий по отношении к остальным большой палец ноги, характеризующийся изогнутостью внутрь;
- Дефект в шейном отделе позвоночника;
- Короткий большой палец рук;
- Отставание в развитии;
- Глухота;
- Облысение;
- Искривление пятого пальца ног;
- Кривошея;
- Сколиоз;
- Тугая подвижность костного сочленения.
Известны случаи, когда уплотнения самостоятельно рассасываются, но в последствии образуются снова. После чего через 2-3 недели происходит их окостенение, в результате которого стает невозможным дальнейшее их рассасывание. У взрослого пациента прогрессирующая болезнь Мюнхеймера приводит к слиянию десятков очагов. Что в последствии вызывает глубокую инвалидизацию и летальный исход.

Диагностика
Диагностирование оссифицирующей фибродисплазии возможно после окончания форсированного костеобразования, которое происходит с момента рождения до 14 лет. Часто заболевание обнаруживается спустя 2 года после его развития. Диагностирование проводится следующими методами:
- Внешним осмотром;
- Рентгенографией;
- Генетической консультацией.
Во время внешнего осмотра у детей до 10-летнего возраста обнаруживаются многочисленные уплотнения, располагающиеся в разных частях тела. В более старшем возрасте определяется пониженная суставная подвижность, искривленный позвоночник, гетеротропические оссификационные очаги. Уплотнения можно увидеть даже невооруженным глазом.
В исключительных случаях может потребоваться гистологическое исследование, но оно проводится редко, чтобы исключить появления новых уплотнений.
Рентгенография полезна на начальном этапе оссифицирующе патологии, поскольку позволяет выявить дисплазию кости, клинодактилию. На поздних стадиях костные уплотнения появляются в мышечной ткани, сухожилиях. Если болезнь зашла далеко, то на рентгеновском снимке не возможно визуализировать внутренние органы.

Лечение
На сегодняшний день не существует методов, вылечивающих прогрессирующую оссифицирующую фибродисплазию. Но американскими учеными активно проводятся исследования гена, который влияет на развитие патологии. Разрабатываются генные блокаторы, которые способны остановить процесс мутации. Восстанавливается нормальное синтезирование белков за счет нейтрализации мутировавшего гена.
Для лечения оссифицирующей фибродисплазии используют следующие лекарственные препараты:
- Этидронат, который замедляет формирование костной ткани. Но ударная доза средства не только оказывает воздействие на патологические кости, но и приводит к разрушению основного скелета;
- Лекарственные средства, основанные на Ламидроновой кислоте, схожи своим действием с предыдущими препаратами, но не так активно влияют на основные кости;
- Преднизолон оказывает мощное противовоспалительное действие, снижает боль, устраняет опухоли. Курс лечения данным средством не должен превышать 2 недель. Иначе происходит ускорение роста костной ткани, подавление иммунитета, ухудшение зрения;
- Целебрекс замедляет воспаление;
- Метаксалон используется для снижения вспышек заболевания, приводит к ослаблению спазмов.
Улучшает состояние больных, страдающих от фибродисплазии, иммунотерапия Интерфероном. Ее действие направлено на замедление прогресса патологии, уменьшение числа уплотнений. Для облегчения самочувствия пациентам назначают обезболивающие и седативные препараты.
Наибольшую безопасность для больных представляет оперативное вмешательство по поводу устранения уплотнений и костного разрастания. После процедуры обнаруживается усиление процесса окостенения.
На начальном этапе оссифицирующей патологии больному назначается электрофорез на основе йодида калия. Противопоказаны пациентам УВЧ, массажи, парафинотерапия.
Людям с оссифицирующей фибродисплазией необходимо исключение хирургического вмешательства, любых видов инъекций для минимизации риска возникновения новых травм.
Данное заболевание имеет неблагоприятный исход. На продолжительность жизни больных оказывают влияние создание условий, исключающих травм, увечий, инфекционных заболеваний.
Клинические и рентгенологические проявления оссифицирующей прогрессирующей фибродисплазии у детей
Прогрессирующая оссифицирующая фибродисплазия (ФОП), оссифицирующий прогрессирующий миозит (ОПМ), болезнь Мюнхмайера, болезнь «второго скелета» — редкое, врожденное, инвалидизирующее заболевание с встречаемостью 1 на 2 млн человек.
Прогрессирующая оссифицирующая фибродисплазия (ФОП), оссифицирующий прогрессирующий миозит (ОПМ), болезнь Мюнхмайера, болезнь «второго скелета» — редкое, врожденное, инвалидизирующее заболевание с встречаемостью 1 на 2 млн человек.
Фибродисплазия характеризуется врожденной костной патологией и прогрессирующим гетеротопическим окостенением мышц, сухожилий, связок, апоневрозов [1, 2]. Нет расовой, половой или географической предрасположенности. Заболевание чаще возникает как результат спонтанной новой мутации. Наследственная передача — аутосомно-доминантная, возможен материнский мозаицизм.
В 2006 г. была обнаружена преимущественно одна и та же гетерозиготная мутация (c.617G>A; p.R206H) в глициновом и сериновом остатке (GS) области активации рецептора 1-го типа активина А (ACVR1)/активиноподобной киназы 2 (ALK2) рецептора 1-го типа костного морфогенетического протеина (КМП), расцененная как генетическая причина ФОП. Семь дополнительных мутаций были идентифицированы впоследствии в GS-области и области киназы ACVR1 у пациентов с «нетипичными» формами болезни [3, 4].
Мутация c.617G>A приводит к замене аргинина на гистидин в кодоне 206 (p.R206H). Структурное гомологичное моделирование белков предсказывает, что замена аминокислоты приводит к конформационным изменениям рецептора, что вызывает изменение его чувствительности и активности [3].
При ФОП предполагается активная выработка КМП-4 сверх порога, что объясняет избыточную оссификацию и эктопическое образование кости постнатально [4].
Врожденные фенотипы, вызванные ФОП-метаморфозом, включают ряд врожденных уродств (деформаций) и скелетных аномалий: деформированный большой палец ноги (основной диагностический признак болезни), дисплазию метафизов, короткие фаланги, короткие большие пальцы рук, синостозы — симфалангизм пальцев, сращение поверхностей шейных суставов, сращение реберно-позвоночных суставов, проксимальные медиальные берцовые остеохондромы, короткие широкие шейки плечевой кости, редкие волосы, глухоту и др. [1, 4, 5].
В отечественной литературе имеются единичные работы о развитии ФОП у детей преимущественно в школьном возрасте [6–8].
В данном сообщении мы представляем наиболее типичные клинические и рентгенологические проявления ФОП у 30 детей в возрасте от 1,5 до 14 лет, наблюдавшихся в клинике детских болезней Первого МГМУ им. И. М. Сеченова в 1968–2010 гг. и два наблюдения с выявленным геном ФОП.
Дети поступали с жалобами на появление на голове мягких эластичных образований, которые постепенно распространялись вниз и уплотнялись. Начало болезни нередко сопровождалось недомоганием, субфебрилитетом, болезненностью очагов, постепенным их уплотнением, нарастанием общей скованности и контрактурами в области крупных суставов. Быстрота распространения процесса колебалась от 2–3 месяцев до 3–5 лет.
У отдельных пациентов наблюдали самопроизвольное исчезновение и/или рецидивы очагов, у других процесс неуклонно прогрессировал. Постепенно дети как бы окутывались костным панцирем, «вторым скелетом», становились малоподвижными, с трудом самостоятельно одевались, принимали пищу, меняли положение в пространстве и в итоге становились инвалидами, зависящими от помощи взрослых (рис. 1, 2). Дети дошкольного возраста могли отставать от сверстников в развитии, старшие были более замкнутыми, но неплохо успевали в школе.
.jpg)
Около половины детей имели типичный признак «классического» варианта ФОП: патологию первых пальцев стоп: укорочение, подвывихи, симфалангизм, монофалангизм и др.
.jpg)
На рентгенограммах обнаруживали внескелетные, костной плотности тени (оссификаты), как единичные, так и сливающиеся между собой в обширные конгломераты. Обычно определялись множественные линейные тени костной плотности с локализацией в мягких тканях конечностей и туловища. Нередко тени переплетались между собой наподобие ветвей дерева. Наиболее частой локализацией были боковые поверхности грудной клетки и внутренние поверхности плеч с образованием синостозов между собой в виде «моста» (рис. 3). У детей младшего возраста эта локализация была первичной. Нередкими были также единичные или множественные экзостозы.
.jpg)
Основные лабораторные параметры, включая общие и биохимические анализы крови и мочи (в т. ч. ферменты и показатели минерального обмена), уровень гормонов щитовидной и паращитовидных желез, оставались нормальными.
Лечение проводили кортикостероидными гормонами (преднизолоном или метилпреднизолоном в дозе 0,5–1 мг/кг массы в сутки) короткими курсами, нестероидными противовоспалительными препаратами (НПВП), внутривенным введением 5% раствора динатриевой соли этилендиаминтетрауксусной кислоты (Na2 ЭДТА), 10% раствора Ксидифона внутрь и/или местно путем электрофореза или мази с Ксидифоном. В последние годы использовали бисфосфонаты нового поколения (динатрия памидронат или Аредиа, Бонефос, Бонвива и др.), антилейкотриеновые препараты (монтелукаст) и блокаторы мастоцитов (кромолин натрия).
Приводим представляющие интерес наблюдения двух пациентов, обследованных генетически в 2010 году.
Артем Т. в 14 лет обратился в клинику в июне 2010 года.
Беременность у матери протекала нормально, но в конце появились отеки ног, А/Д — 150/100 мм рт. ст, прибавка веса 10 кг. Из-за запрокидывания головки ребенка проведено кесарево сечение (12-часовой безводный период). Вес при рождении 3200 граммов, длина 53 см, окружность головки 36 см. Рос и развивался удовлетворительно. Сидит с 6 месяцев, начал ходить с 10 месяцев, первый зуб прорезался в 4 месяца. Вес в один год 11 кг, речь с 1 года 3 месяцев. Грудное вскармливание 2 месяца, далее искусственное, диагностирована анемия легкой степени.
На все прививки, кроме реакции Манту, отмечали подъем температуры до 39–40 °C. При проведении АКДС распухала ягодица (как «стеклянная»).
Перенесенные болезни до школы — ветряная оспа, мононуклеоз, позже — ОРВИ.
В два года поставлен диагноз врожденная вальгусная деформация больших пальцев стоп, сделана операция выправления подвывиха первого пальца левой ноги (вставлена спица).
В 5 лет — диагноз болезнь Пертеса, экзостозная болезнь.
В 6 лет сделана операция по удалению «наростов» (экзостозов) с обеих сторон на задней поверхности коленных суставов. Но возник рецидив, и появились новые очаги спереди.
В 9 лет из-за ушиба (падение с велосипеда) стала плохо сгибаться в колене левая нога, стал прихрамывать.
С 11 лет выявлена двухсторонняя нейросенсорная тугоухость.
В 14 лет (апрель 2010 г.) появилось уплотнение на шее слева. На операции в кивательной мышце выявлен тяж хрящевой плотности. Материал биопсии консультирован в РОНЦ АМН РФ: данных за опухоль нет.
Консультирован в ЦИТО в мае 2010 г., установлены экзостозы и ограничение движений в левой кивательной мышце.
УЗИ левой кивательной мышцы (21.05.2010) — в средних ее отделах определяется участок измененной структуры, захватывающий всю толщу мышцы на протяжении 5,2 см с уплотнением, понижением эхогенности, не имеющий характерной волокнистой структуры; выше и ниже в мышечной ткани есть участки гиперэхогенных мышечных включений (послеоперационные изменения на фоне специфического миозита). Справа — аналогичные включения.
УЗИ передней поверхности правого бедра — на уровне верхней трети — оссификат (2,8 × 0,3 см толщины), в мягких тканях спины справа, на уровне нижних грудных позвонков — оссификат, аналогичный по структуре.
По завершении обследования и очной консультации в РОНЦ им. Н. Н. Блохина в мае 2010 года заподозрен прогрессирующий оссифицирующий миозит.
На консультации генетика установлен диагноз — «фибродисплазия прогрессирующая оссифицирующая с аутосомно-доминантным типом наследования в родословной «de novo». В фенотипе — уплотнение мышц в области шеи, спины, правого бедра, с 9 лет костно-хрящевые экзостозы в верхней трети большеберцовой кости и нижней трети левого бедра, врожденная аномалия первых пальцев кистей и стоп (укорочение, тугоподвижность в межфаланговых суставах), укорочение и расширение шейки бедренных костей.
В клинике детских болезней обследован амбулаторно (15–17.06.2010 г.).
При осмотре мальчик хорошего роста, повышенного питания (рост 160 см, вес 60 кг). Жалуется на затруднение при поворотах шеи и наклонах туловища вперед. Ощущает неловкость в спине и тазе при ходьбе и ощупывании, прихрамывает на правую ногу.
Кожа чистая со следами загара. Укорочение больших пальцев стоп и кистей с подвывихом на ногах — halux valgus (рис. 4). На шее в области левой кивательной мышцы уплотнение с рубцом в центре. Поворот влево затруднен. Наклон туловища вперед лишь до горизонтального уровня. Справа на передней поверхности бедра — уплотнение в толще мышцы. На спине справа на уровне 6-го грудного позвонка определяется костный тяж длиной около 5–7 см, вне связи с позвоночником. Лимфатические узлы не увеличены. Со стороны легких, сердца, органов брюшной полости без патологии.
На R-грамме стоп: справа — вальгусная деформация первого пальца, основная фаланга значительно укорочена, утолщена, смещена латерально (подвывих). Слева — деформация головки плюсневой кости и проксимальной фаланги первого пальца (после операции). Суставная щель неравномерно сужена (рис. 4).
.jpg)
На рентгенограмме тазобедренных суставов с захватом 2/3 бедренных костей — головки бедренных костей уплощены, левая смещена латерально. Слева крыша вертлужной впадины скошена. Утолщение кортикального слоя в бедренных костях. В проекции шейки и верхней половины диафиза бедренной кости слева в мягких тканях определяются тени костной плотности. Справа — единичные тени в проекции шейки бедренной кости (рис. 5).
На рентгенограммах кистей соотношение костей не нарушено. Отмечается метаэпифизарный остеопороз. Пястные кости укорочены. Слева основная фаланга первого пальца укорочена, гипоплазия эпифиза. Средние фаланги V пальцев с искривленностью.
ЭхоКГ (17.06.2010 г). Размеры полостей сердца, толщина миокарда в пределах нормы. Систолическая и диастолическая функции левого желудочка не нарушены. Давление в легочной артерии в норме. По передней стенке правого желудочка минимальное расхождение листков перикарда в диастолу 1,3–1,5 мм. МАРС: добавочные базальные хорды и трабекула в полости левого желудочка. Удлинен евстахиев клапан.
Клинический диагноз: «Фибродисплазия оссифицирующая прогрессирующая. Классическая форма. Поздняя стадия. Врожденные аномалии больших пальцев стоп: укорочение и утолщение основных фаланг с подвывихом (Halux valgus) и кистей (укорочение пястных костей и основной фаланги первого пальца слева с гипоплазией эпифиза. Искривление средних фаланг V пальцев). Костно-хрящевые экзостозы слева на уровне нижней и верхней третей бедренной кости. Укорочение и расширение шейки бедренных костей. Множественные внескелетные костные образования (оссификаты) в толще мышц спины, бедер, шеи.
МАРС: добавочные базальные хорды и трабекула в полости левого желудочка, удлинение евстахиевого клапана. Двусторонняя нейросенсорная тугоухость.
В лаборатории ДНК-диагностики: найдена мутация Arg206 His в гене ACVR1».
Заключение. Ребенок наблюдался длительно с диагнозами врожденной вальгусной деформации больших пальцев стоп, экзостозной болезни, болезни Пертеса, подозрением на опухоль шеи с биопсией измененного участка кивательной мышцы; получал все прививки и вакцинации, несмотря на высокую лихорадку на каждое введение вакцин, не соблюдал необходимого щадящего двигательного режима, многократно травмировался, подвергался многочисленным лучевым исследованиям, операциям и лишь через 14 лет диагноз ФОП верифицирован и подтвержден генетически.
В настоящее время ему рекомендованы щадящий двигательный режим, отвод от всех внутримышечных инъекций, манипуляций в области нижней челюсти, профилактика ОРВИ (Арбидол, санация полости носа — промывание морской водой, капли — Маример, вакцинация при эпидемии гриппа подкожно или интраназально), плавание в бассейне с морской водой или в море, применение преднизолона (2 мг/кг 5–7 дней) и бисфосфонатов (Аредиа) при возникновении новых оссификатов, оформление инвалидности и динамическое наблюдение в специализированном стационаре.
Второй пациент — Алеша О. под наблюдением клиники с 1 года 3 месяцев.
В анамнезе: мать 27 лет, здорова, отец 39 лет, страдает бронхиальной астмой, алкогольной зависимостью. На 18-й неделе беременности мать перенесла грипп, в последующем из-за непропорционального развития головки была госпитализирована в отделение патологии беременности. Роды в срок, физиологические. Вес ребенка 2530 г, длина — 48 см. Оценка по шкале Апгар 8–9. Закричал сразу. Кормление грудью 8 месяцев, отмечалась гипогалактия. С раннего детства отмечено «плохое» держание головки в положении на животе, в 3 месяца — массаж из-за гипертонуса мышц шеи. Сидит с 7 месяцев, ходит с 12 месяцев, зубы с 5 месяцев.
В возрасте 5 месяцев упал с дивана без последствий. Позже в 12 месяцев повторное падение, ударился лбом. На месте ушиба возник отек размером с куриное яйцо. Получал гидрокортизон, примочки местно с эффектом.
При рентгенографии и томографии выявлена врожденная аномалия С1–С2 — деформация тел позвонков в виде платиспондилии, незаращение на уровне задних отделов (spina difida posterior), гипоплазия зубовидного отростка С2.
При ЭхоКГ размеры камер сердца выше возрастной нормы (мальчик повышенного питания: вес в 1 год 2 месяца 15 кг). Дополнительная хорда в левом желудочке.
При первичном осмотре ребенок хорошего физического развития, повышенного питания, мобилен, общителен. Имеется «скованность» и приподнятость в плечевом поясе, напряженность и уплотнение мышц шеи, ограничение в поворотах головы и наклоне назад. Движения в суставах конечностей не ограничены, деформаций нет. Приседает, ходит, бегает свободно. Кожные покровы и слизистые чистые. Зубы 10/8. По внутренним органам — без патологии, выявлена гипоспадия.
Имеются диагностические признаки классической формы ФОП — укороченные с подвывихом большие пальцы стоп (рис. 6), а также врожденная патология шейных позвонков и гипоспадия.
.jpg)
Общие анализы крови и мочи, биохимическое исследование крови в пределах нормы.
Генетическое обследование выявило наличие мутации Arg206His в гене ACVR1, характерное для классического варианта ФОП.
Клинический диагноз: «Фибродисплазия оссифицирующая прогрессирующая. Классическая форма. Ранняя стадия. Начальная фаза болезни. Рецидивирующие опухолеподобные образования на голове и в области шеи. Хрящеподобные элементы в виде продольных эластичных тяжей (теней на рентгенограммах) в мягких тканях надплечий и вдоль позвоночника. Врожденные костные аномалии: укорочение больших пальцев стоп. Недоразвитие зубовидного отростка 2-го шейного позвонка. Платиспондилия и незаращение задних отделов тел С1–С2. Гипоспадия. Мутация Arg206 His в гене ACVR1».
Ребенок наблюдается в течение года. Заболевание не прогрессирует (рис. 7). Периодически на голове при легких травмах возникают эластичные «опухоли», быстро исчезающие при применении мази с Ксидифоном. Дан отвод от всех внутримышечных прививок и вакцинаций. Рекомендован охранительный двигательный домашний режим. При падениях и образовании мягких опухолей на голове — мазь с Ксидифоном, внутрь НПВП, антилейкотриены. Динамическое наблюдение в клинике для коррекции терапии. Оформление инвалидности детства.
Таким образом, наши наблюдения свидетельствуют о недостаточном знакомстве с ФОП врачей различных специальностей — педиатров, хирургов, ортопедов, что приводит к длительной верификации диагноза, ненужным вмешательствам и травмам мягких тканей (операции, биопсии, внутримышечное введение лекарств, вакцинации).
Обобщая литературные и собственные данные, следует считать для диагноза главным наличие врожденной патологии больших пальцев стоп (нередко в сочетании с патологией пальцев кистей) и внескелетные оссификаты. У детей дошкольного возраста, как правило, они возникают на шее, плечевом поясе и постепенно распространяются вниз. У старших детей оссификаты могут появляться в любых участках мягких тканей (мышцах, сухожилиях и др.), чаще вследствие травм любого генеза или на фоне гриппа и ОРВИ. При подозрении на ФОП необходим поиск мутаций в гене ACVR1.
К сожалению, прогноз болезни неудовлетворительный. Очень важно соблюдать все профилактические меры для избежания любых травм мягких тканей и проводить профилактику ОРВИ и гриппа, которые могут ускорить прогрессирование болезни и спровоцировать легочно-сердечную недостаточность.
В настоящее время содружеством ученых активно разрабатываются и апробируются (в эксперименте и на животных) препараты, обладающие способностью блокировать мутации в гене ACVR1, что позволит предупредить или прервать гетерогенную оссификацию и улучшить состояние пациентов.
Литература
- Mc Kusick V. A. Heritable Disorders in Connective Tissue. St. Louis C. V. Mosby (pub) (4 th ed.), 1972.
- Kaplan F. S., Le Merrer M., Glaser D. L., Pignolo R. J., Goldsby R. E., Kitterman J. A., Groppe J., Shore E. M. Fibrodysplasia ossificans progressiva // Best Pract Res Clin Rheumatol. 2008, 22: 191–205.
- Shore E. M., Xu M., Fe >Т. В. Рябова,кандидат медицинских наук
Н. А. Геппе, доктор медицинских наук, профессор
Г. В. Михалева
И. Г. Сермягина
Первый МГМУ им. И. М. Сеченова, Медико-генетический научный центр РАМН, Москва
Фибродисплазия: как дети превращаются в камень

Фибродисплазия оссифицирующая прогрессирующая или ФОП (от лат. fibro — волокно, dis — расстройство, нарушение, plasis — строение, структура и os, ossis — кость, facio — делать; окостенение) «мягкая соединительная ткань, которая прогрессивно превращается в кость». Очень редкое и тяжелое по своему течению генетическое заболевание, при котором мышцы, сухожилия и связки постепенно превращаются в кости. Процесс прогрессирует с годами, начинаясь обычно в пределах десятилетнего возраста у детей с мутацией определенного гена.
Эта странная, пугающая болезнь известна уже не первое столетие. Но до сих пор не найдено эффективного лекарства или метода лечения недуга, заставляющего тело превращаться в кость, сковывающего его и приводящего в конце концов к гибели больного.
Рассказывает о фибродисплазии, известной также, как «синдром каменного тела».
Каменеющие дети: история
Первые сведения о детях, в теле которых образовывались кости вместо мягких тканей, относятся к XVII веку. Записки о наблюдениях за каменеющим пациентом оставил врач из Франции Ги Пэтин. Пэтин признавал, что встречается с такой патологией впервые, и попытался провести аналогию с другим случаем из своей практики — с так называемым литопедионом, или плодом замершей беременности, подвергшимся кальцификации в утробе матери. Но на самом деле это совершенно разные патологии, не имеющие ничего общего.
На полвека позднее англичанин Джон Фрек описывал мальчика-подростка, у которого вдоль позвоночника образовывались твердые кораллообразные наросты. Подросток пришел к доктору, потому что Фрек разрабатывал методы лечения желчнокаменной и мочекаменной болезней, рассудив, что каменные наросты на его спине врач тоже сможет убрать.
К сожалению, помочь мальчику Фрек не мог, ему оставалось только наблюдать, как странная болезнь быстро прогрессирует.
В середине XVIII века был описан еще один случай фибродисплазии. Болезнь проявилась у 18-летнего юноши и унесла его жизнь в 60 лет. Предположительно, больной умер от голода, так как его челюсти перестали открываться. Описание его скелета напоминает фильмы ужасов: все кости бедняги превратились в жесткую структуру, утратившую способность сгибаться и двигаться. Спину закрывали сросшиеся в одну пластину лопатки, кости конечностей стали более похожи на ветви кораллов, а на пятках торчали костяные «шпоры».
В настоящее время заболевание по-прежнему считается очень редким: на Земле проживает всего около 600 людей, страдающих этой патологией. И редкость заболевания замедляет поиски средств для его лечения: ученым не хватает информации. Хотя удалось уже выяснить кое-что о природе болезни.
Кости вместо мышц: почему тело костенеет
Что известно сегодня о заболевании, известном как фибродисплазия оссифицирующая прогрессирующая (ФОП)?
Болезнь может проявляться в результате спонтанной мутации или наследоваться по аутосомно-доминантному типу. Сами по себе изменения, происходящие в теле больного, не смертельны, но прогрессирование болезни в какой-то момент приводит к возникновению настоящих оков для тела, и жизнь прекращается. Это может быть, например, полное окостенение ребер, которые теряют способность двигаться, и больной просто больше не может дышать.
Механизм окостенения достаточно сложен. Известно, что причиной роста костей в неположенных местах становится сбой в иммунной системе. В травмированной или поврежденной мышце высвобождаются специфические белки — цитокины. В ответ на появление цитокинов эндотелий кровеносных сосудов вырабатывает мезенхимальные стволовые клетки, которые и выполняют «ремонт» поврежденного участка.
При повреждении гена ACVR1 стволовые клетки разделяются на остеобласты и хондроциты. Остеобласты, которые в норме образуют кости скелета, при генетическом сбое могут начать формировать костную ткань не там, где она и должна быть, а в месте повреждения мягкой ткани. И тогда в поврежденной мышце образуется совершенно ненужная кость.
Попытки удалить новообразования костной природы приводят к усугублению проблемы, так как любая операция дополнительно травмирует ткани, вызывает повышенное высвобождение цитокинов, и процесс ускоряется. Окостенение может затрагивать не только мышцы, но и соединительную ткань — сухожилия и связки.
При длительном течении болезни в организме словно формируется второй скелет поверх естественного, нормального. Это хорошо видно на костных структурах больных, завещавших свое тело науке: они могут стоять, не рассыпаясь и не сгибаясь за счет того, что «второй скелет» образовал с первым прочную, несгибаемую конструкцию.
Одним из таких больных был Гарри Истлэк, родившийся в 1933 году. Первые признаки болезни появились уже в пятилетнем возрасте. Врачи делали все, что могли, но Гарри становилось только хуже. Ему вырезали странные костные образования — а на их месте вырастали еще более крупные, и чем больше было операций, тем сильнее Гарри страдал от прогрессирующего окаменения. Тогда было еще неизвестно, что окостенение усиливается от любого повреждения тканей…
Истлэк завещал свое тело ученым, чтобы дать им материал для изучения недуга. Прожил он всего 40 лет и умер от болезни сердца.
Заболевание может проявляться в раннем детстве, но это не обязательно. Иногда ФОП дает о себе знать в 15-19 лет. Одним из заметных признаков фибродисплазии является врожденная патология больших пальцев на ногах: они укорочены и повернуты внутрь под неестественным углом.
Типичное начало болезни затрагивает область вдоль позвоночника и шеи. В прилегающих мышцах могут начать появляться пластины и ленты кальцифицированной ткани. Не изменяются всего несколько мышц тела: сердце, гладкая мускулатура кишечника, диафрагма и язык.
Болезнь, приводящая к инвалидности — фибродисплазия (болезнь Мюнхеймера)
В этой статье мы расскажем вам о тяжелейшем инвалидизирующем заболевании, которое встречается очень редко — 1 человек на 2 млн населения. Всего в мире насчитывается около 500 больных этим недугом. Он заключается в том, что мышцы больных постепенно превращаются в кость, а сам человек постепенно, по сути, становится статуей и утрачивает возможность двигаться. Название этого недуга — фибродисплазия.
Давайте узнаем, из-за чего развивается эта болезнь, кто ей подвержен и реально ли ее вылечить.
Что такое фибродисплазия?
Это болезнь, спровоцированная изменением гена ACVR1, которая проявляется наследственными дефектами развития. Этот недуг получил также название «болезнь второго скелета». Прогрессирующая оссифицирующая фибродисплазия (ПОФ, болезнь Мюнхеймера) провоцируется воспалениями в сухожилиях, мышцах, связках, мягких тканях, которые с течением времени костенеют. Говоря проще, ушибы и даже небольшие ранки не поддаются лечению и на их месте появляются костные образования, по этой причине недуг получил еще одно название «болезнь второго скелета».
У новорожденных можно с 95% вероятностью определить ПОФ — большой палец ноги загнут внутрь и зачастую там отсутствует сустав. Обычно болезнь проявляет себя в течение первых 10 лет жизни ребенка и совершенно внезапно. У детей все начинается с шеи и постепенно спускается до нижних конечностей.
Скорость развития также невозможно установить точно, но чем больше травм получает больной и чем больше хирургических вмешательств происходит, тем быстрее развивается недуг.
Довольно часто ПОФ путают с раком и прочими болезнями, в результате которых могут развиться подобные костные образования. Врачи удаляют эти образования, но, к сожалению, это приводит к усугублению ситуации и ускорению развития фибродисплазии.
В этой передаче рассказывается о двух больных девушках (русской и американки). Они рассказывают о том, как все началось и возможно ли вылечить ПОФ. У американской девушки врачи первоначально диагностировали рак и ампутировали больную руку в раннем детстве, и только потом выяснилось, чем она в действительности больна.
Причины
Что именно запускает мутацию на генном уровне, ученые пока не могут выяснить. На данный момент известно только то, что это наследственный недуг и передается аутосомно-доминантным путем. Это значит, что у больного ПОВ 100% будут дети с точно такой же патологией. Однако люди с болезнью Мюнхеймера редко доживают до возраста, когда могут рожать детей. А если и доживают, то женщинам смертельно опасно беременеть из-за изменений, происходящих в организме. Так что ПОФ является итогом индивидуальных генетических мутаций.
Только в 2006 году ученые выявили, мутация какого именно гена причастна к развитию фибродисплазии и сейчас проходят исследования, чтобы создать препарат, позволяющий лечить этот недуг.
Зачастую у носителей мутировавшего гена недуг никак не показывает себя до какого-то времени. Толчком к развитию могут послужить:
- травмы,
- вакцинации,
- вирусное заражение,
- хирургические вмешательства.
Симптоматика
Самыми очевидными симптомами болезни Мюнхеймера является окостенение мягких тканей, но кроме этого могут быть и другие дефекты, указывающие на возможное наличие недуга:
- короткий изогнутый внутрь большой палец стопы,
- дефекты в позвоночнике, особенно в шейном отделе,
- короткий большой палец на руках,
- возможно облысение, глухота, замедленное развитие,
- искривление или скос пятого пальца.
Достаточно часто ПОФ провоцирует развитие кривошеи, сколиоза и тугой подвижности костных сочленений.
Если вы заметили у ребенка подобные признаки недуга, то обратитесь к врачу. Доктор, если посчитает нужным, назначит генетическое исследование, в результате которого либо оправдаются подозрения на наличие генной мутации, либо нет.
Диагностика и лечение
Как мы уже упоминали выше, зачастую на ранней стадии фибродисплазии врачи ставят неверный диагноз, что в свою очередь только усугубляет развитие недуга. Обычно болезнь Мюнхеймера диагностируют в возрасте около 14 лет, когда все симптомы становятся явными.
На рентгене видно, что существуют костные образования в области шеи, груди, а с возрастом эти образования наблюдаются и в других местах.
Генетический анализ — единственный способ точно определить наличие изменений в ДНК.
К сожалению, пока болезнь Мюнхеймера считается неизлечимой. Однако американские ученые, проводя исследования, уже выявили, как мы говорили, ген ACVR1, при мутации которого происходит развитие недуга. Сейчас продолжается работа над генными блокаторами, которые должны остановить мутацию. Из-за того, что это заболевание очень редкое довольно сложно проводить исследования.
Несмотря на это, в 2011 году была разработана особая РНК-молекула, которая может убрать поврежденную копию гена ACVR1, никак не влияя на его нормальную копию.
Чтобы проверить, насколько эффективен данный способ, были проведены проверки на стволовых клетках, которые были получены из молочных зубов больных фибродисплазией. В клетках были измененные и нормальные копии ACVR1/ALK2 рецептор-белков. В ходе опыта ученые выяснили, что созданная мРНК способна восстанавливать нормальный синтез белков такой же, как у здоровых людей, нейтрализовав мутировавший ген.
В этом видео рассказывается о том, каким образом проводятся эти анализы и как в принципе выявляются генетические заболевания.
Еще 10 лет назад было невозможно представить, что этот недуг излечим. Сейчас у страдающих болезнью второго скелета появилась надежда на здоровую и счастливую жизнь.
Конечно, предстоит еще большая работа, этот подход к лечению нужно опробовать на лабораторных мышах и только потом перейти к испытаниям на людях. Но, тем не менее, это уже большой шаг. Будем надеяться, что исследования закончатся успешно.
Прогрессирующая оссифицирующая фибродисплазия фоп
Дети, рожденные с Фибродисплазией, отличаются характерной патологией большого пальца ноги – одна или несколько фаланг пальца искривлены вовнутрь и иногда в нем не хватает сустава. Этот палец и дает 95 % – ную вероятность заболевания у ребенка. Также болезнь характеризуется обострениями, как правило, без видимых причин. Обострения бывают нескольких видов, самое распространенное — появление под кожей ребенка уплотнений размером от одного до десяти сантиметров неопределимого характера, на шее, спине, предплечьях в возрасте до 10 лет, в любом другом месте в более старшем возрасте. Одним из признаков является отек мягких тканей головы, как при незначительном повреждении (ушиб, укус насекомого, царапина), так и при сильных травмах. Отек не спадает до месяца, не реагирует ни на какую медикаментозную терапию.На месте уплотнений могут возникать оссификации, она не нуждается ни в каком лечении и не поддается никаким препаратам. Болезнь часто путают с раком и другими заболеваниями, подобные затвердения пытаются удалять, что приводит к ещё более бурному росту «лишних» костей,и является основной причиной инвалидизации страдающих этим заболеванием на данный момент.
Лечение
Вспышки ФОП являются спорадическими и непредсказуемыми, и есть огромная разница в каждом индивидуальном проявлении. Несколько базовых научных исследований подтвердили невозможность предсказать начало, силу, последствия и продолжительность обострения ФОП, хотя особенности анатомических патологий уже были описаны. Редкость ФОП и ее непредсказуемая природа делает чрезвычайно трудным оценку эффективности любого терапевтического вмешательства, факт, признанный 1918 году врачом Джулиус Розенстирн :
«Болезнь развивается со всеми видами средств и альтернатив дефектного метаболизма; каждое отдельное терапевтическое вмешательство с более или менее отмеченным успехом, наблюдаемым исключительно его оригинальным автором, в будущем раскритиковывается любым последователем. Во многих случаях, признаки болезни исчезают часто спонтанно, таким образом, терапевтический эффект (любого лечения) не может быть полностью подтвержден своей эффективностью.»
Эти слова звучат правдоподобно и сегодня, несмотря на то, что они были написаны почти столетие назад. В настоящее время не существует никаких доказанных эффективных методов профилактики или лечения ФОП. С открытием гена ФОП стало возможным понимание патогенеза заболевания. Перспективной является непосредственная работа с геномом, но в настоящее время данные методы лечения находятся лишь на стадии предварительных лабораторных исследований и не применяются в клинической практике.
Генетический аспект и будущее лечение
В 2006 году исследовательская группа из Университета штата Пенсильвания открыла ген, мутация в котором приводит к этому заболеванию. Чтобы идентифицировать хромосомное местоположение для гена ФОП, консервативной связки всего генома, анализ проводился с помощью наблюдения за пятью семьями с самыми однозначными особенностями ФОП. Этот подход идентифицировал связку ФОП к хромосоме 2q23-24.
Генотерапия: В настоящее время идет работа над генными блокаторами мутации в гене ACVR1/ALK2.В декабре 2012 завершены с успехом опыты по лечению гетеротопической оссификации лабораторным мышам.Препарат показал свою действенность как в генетически обусловленной так и в посттравматической оссификации. В течение 2013 года начнутся испытания препарата в первой фокус – группе пациентов ФОП. Золотой стандарт для всех исследований нового препарата — тройной слепой перекрестный метод с применением плацебо, но такие исследования будет достаточно трудно провести при ФОП, ввиду малого количества пациентов с болезнью в уже развитой стадии, неоднозначным патогенезом болезни, а также большой разницей в проявлениях обострений у пациентов. Ввиду того, что фибродисплазия является чрезвычайно редким заболеванием, с разной тяжестью проявлений, экспериментальным методам лечения, предстоит пройти серьёзную оценку на предмет дозировки и продолжительности курса.
Ссылки
- [3] – Информационный сайт для страдающих заболеванием и их врачей
- The International FOP Association
- [4]
- [5]
- Rare ‘stone man’ gene that changes muscle into bone -News-World-US & Americas-TimesOnline. Проверено 2 июня 2007.
Frederik Kaplan The medical management of Fibrodysplasia Ossificans Progressiva: current treatment considerations |2008

